fu-msa
Preliminary version
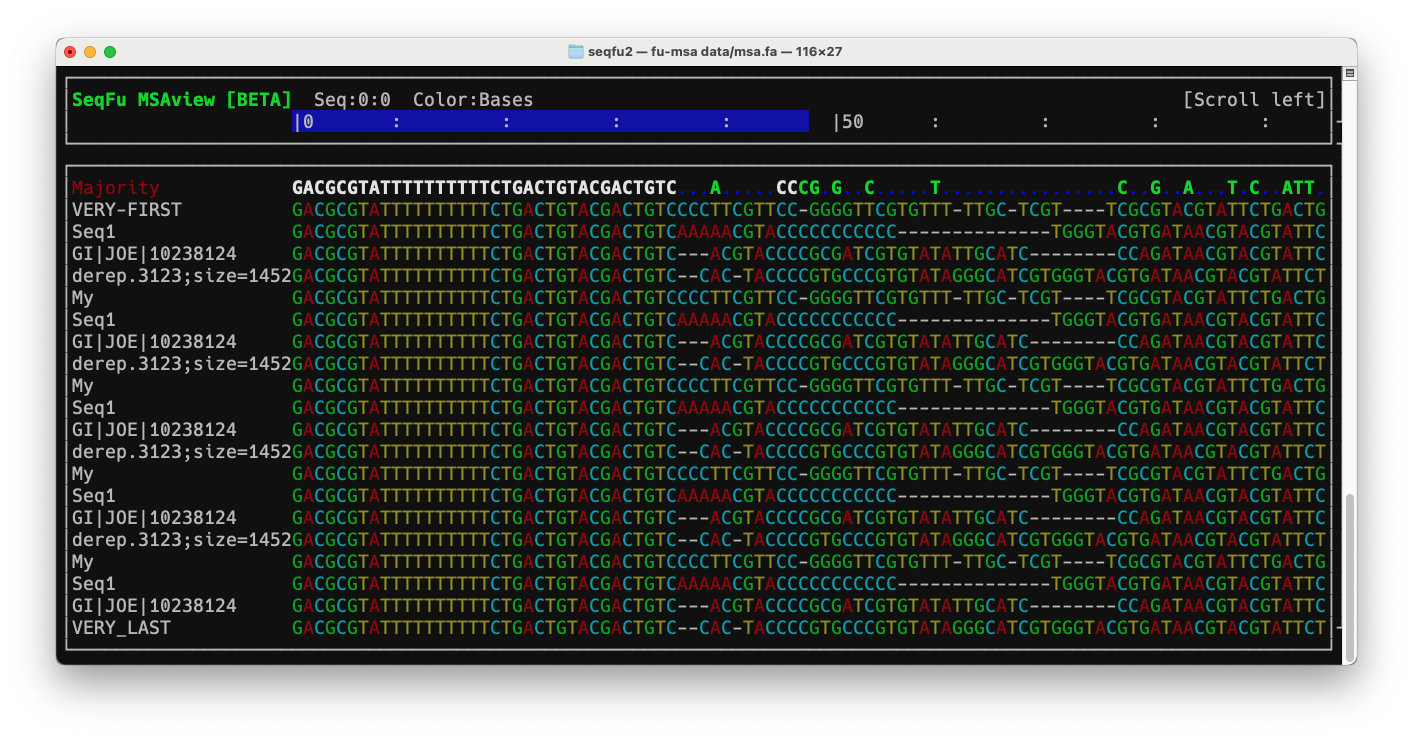
Interactive multiple sequence alignment viewer from the Command Line.
Usage:
full [options] <MSAFILE>
Keys:
Scroll Horizontally Left and Right arrow
By 10 bases L, K
By 100 bases ShiftL, ShiftK
To the beginning 1
To middle parts 2..9
To the end 0
Scroll Vertically A, Up Arrow/ Z, Down Arrow
Jump to top ShiftA, PageUp
Jump to bottom ShiftZ, PageDown
Rotate color scheme Tab
Refresh screen F5
Resize seq labels -,+
Search / (seqname, ":INT", "#SEQ")
Quit Q, CtrlC
Options:
-m, --mouse Enable mouse
-n, --norefresh Disable autorefresh
-j, --jumpsize INT Jump size (big jump is 10X) [default: 10]
Visualization settings:
-i, --seqindex INT Start visualization at this sequence [default: 0]
-p, --seqpos INT Start visualization at this nucleotide [default: 0]
-w, --label-width INT Sequence label width [default: 20]
-s, --setting-string STR Settings string (overrrides all other settings) is in the
format Seq:{seqindex}:{seqpos}:{labelwidth} and is the
return value of the program when it is closed.
More documentation online at https://telatin.github.io/seqfu2/
Keyboard
Horizontal scrolling
- :arrow_left:, :arrow_up: : scroll by one nucleotide
K,L: scroll left and right respectively, by 10 nucleotidesShift + K,Shift + L: scroll left and right respectively, by 100 nucleotidesEnd: Move to sequence endHome: Move to the beginning of the sequence1: Move to the first base2..9: Move to 20% .. 90% of the sequence0: Move to the last base
Vertical scrolling
A,Zand :arrow_up:, :arrow_down: : Move up and down by one sequenceShift + A,Shift + Z: Move to top and move to bottom
Search
/Trigger search, then:- Type the query
- Hit
Enterto submit orEscto abort
- The query can be:
- part of a sequence name
:followed by a sequence index (eg::0to go to the first sequence)#followed by a sequence (eg:#ATTACto jump to the position of the first occurrence of ATTAC)
Visualization
Space: toggle consensus sequence from “Consensus” (show bases identical across all the sequences) to “Majority” (show bases shared by 50% of the sequences or more)Tab: toggle color scheme-,+: Decrease and increase by one the sequence label widthF5,R: refreshF6: toggle autorefresh on/offH: toggle help screen (only major keys are reported)
Mouse
M: toggle mouse on/off- When mouse is on:
- Click in a nucleotide to set that position as first (scroll right)
- Click to the sequence name area (left) to scroll left
Resuming session
When pressing quit (Q) the program will print a configuration string like: Seq:0:6:20 that can be used to resume the session at the same position with:
fu-msa {input_file} --setting-string Seq:0:6:20
The settings string is in the format Seq:{seqindex}:{seqpos}:{labelwidth}
Colors
- DNA:
A(red),C(cyan),G(green),T(yellow) - Protein: “Lesk” scheme:
- Hydrophobic, green
- Small non polar, yellow (should be orange)
- Polar, magenta
- Negative charge, red
- Positive charge, cyan (should be blue)
Screenshot
