seqfu qual
qual allows to detect the range of qualities of a FASTQ file, returning the possible encodings. Can be used to detect the last qualified position ().
Usage: qual [options] [<FASTQ>...]
Quickly check the quality of input files returning the detected encoding
and the profile of quality scores.
To read from STDIN, use - as filename.
-m, --max INT Check the first INT reads [default: 5000]
-l, --maxlen INT Maximum read length [default: 1000]
-k, --skip INT Print one sequence every INT [default: 1]
Qualified position:
-w, --wnd INT Sliding window size [default: 4]
-q, --wnd-qual FLOAT Minimum quality in the sliding window [default: 30.0]
-z, --min-qual FLOAT Stop the sliding windows when quality is below [default: 18.0]
Additional output:
--gc Print GC content as extra column
-p, --profile Quality profile per position (will comment the summary lines)
-c, --colorbars Print graphical average quality profile
Other options:
-v, --verbose Verbose output
-O, --offset INT Quality encoding offset [default: 33]
--help Show this help
Example
Check if a set of files is likely in Illumina 1.8 encoding:
seqfu qual data/primers/*
data/primers/16S_R1.fq.gz 7.0 38.0 Sanger;Illumina-1.8; 33.42+/-8.47 249
data/primers/16S_R2.fq.gz 2.0 38.0 Sanger;Illumina-1.8; 31.96+/-9.53 205
data/primers/16Snano_R1.fq.gz 7.0 38.0 Sanger;Illumina-1.8; 33.37+/-8.63 246
data/primers/16Snano_R2.fq.gz 2.0 38.0 Sanger;Illumina-1.8; 32.05+/-9.54 220
data/primers/art_R1.fq.gz 40.0 40.0 Illumina-1.3;Sanger;Illumina-1.5;Solexa;Illumina-1.8; 40.00+/-0.00 95
data/primers/art_R2.fq.gz 40.0 40.0 Illumina-1.3;Sanger;Illumina-1.5;Solexa;Illumina-1.8; 40.00+/-0.00 93
The artifical datasets (art*) were designed to be compatible with most encodings, while the 16S* files are real Illumina 1.8 sequences.
Output
For each file a tab separated record is printed:
- Filename
- Minimum quality value
- Maximum quality value
- Possible encoding (semicolon separated list)
- Mean, StDev of the quality value
- Last qualified position
- GC content (5 decimal positions) if enabled via
--gc
Per base statistics
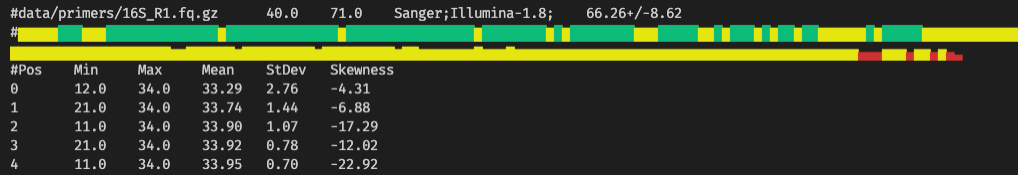
With the --profile option tabular overview of the quality scores per nucleotide position of the read is printed:
#data/primers/16Snano_R1.fq.gz 40.0 71.0 Sanger;Illumina-1.8; 66.37+/-8.63
#Pos Min Max Mean StDev Skewness
0 27.0 34.0 33.95 0.50 -12.36
1 27.0 34.0 33.97 0.46 -14.78
2 11.0 34.0 33.73 2.28 -9.08
3 23.0 34.0 33.92 0.76 -13.09
4 31.0 34.0 33.99 0.20 -15.20
5 28.0 38.0 37.76 0.86 -7.20
6 10.0 38.0 36.26 3.98 -4.89
...
298 7.0 37.0 26.34 10.34 -0.58
299 7.0 37.0 25.62 11.23 -0.55
300 7.0 37.0 21.29 9.26 -0.09
Graphical summary
With the --colorbar option a graphical (Unicode) colored histogram of the average quality per base position is printed after each file.
Use with DADA2
The last column of the tabular report (last qualified position), can be used to automatically detect the truncating position when using DADA2. Dadaist uses SeqFu to automatically detect the qualified region.