There are lots of powerful things you can do with the Markdown editor. If you’ve gotten pretty comfortable with writing in Markdown, then you may enjoy some more advanced tips...
Even the press, the classroom, the platform, and the pulpit in many instances do not give us objective and unbiased truths. To save man from the morass of propaganda, in...
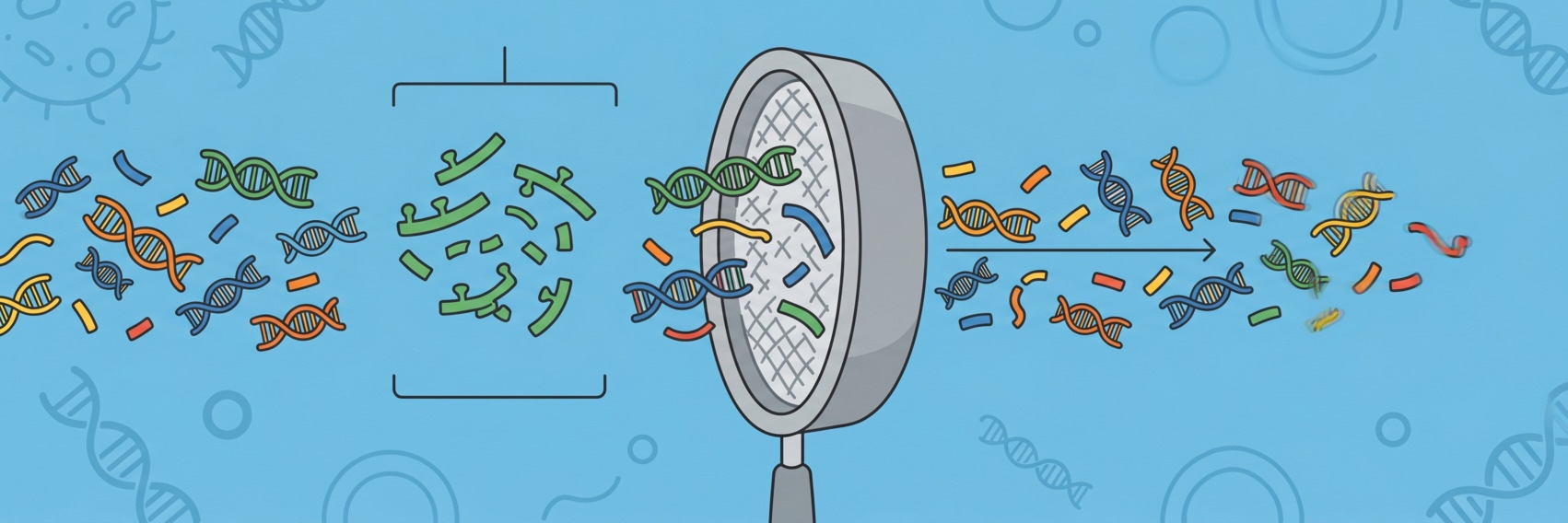
Two popular formats to store structured data are XML and JSON.
They are used everywhere in computer science and appear more and more often in
bioinformatics analyses as well.
Two popular formats to store structured data are XML and JSON.
They are used everywhere in computer science and appear more and more often in
bioinformatics analyses as well.
A very first primer on the Linux command line for bioinformatics. Open your terminal and start typing the first commands! There are several pages dedicated to Bash in this website,...
The idea to check if some condition is true before executing some commands is very simple and useful. The Bash syntax, for historical reasons, is probably the ugliest you’ll ever...
After a small introduction to Bash scripting, we finally create a first bioinformatics script… introducing one of the loops we can use with the shell. A loop is a structure...
After using the Linux terminal for a while, everyone wants to be able to write simple “scripts” to perform repetitive tasks. They looks like recipes, with the main difference of...
A very first primer on the Linux command line for bioinformatics. Open your terminal and start typing the first commands! There are several pages dedicated to Bash in this website,...
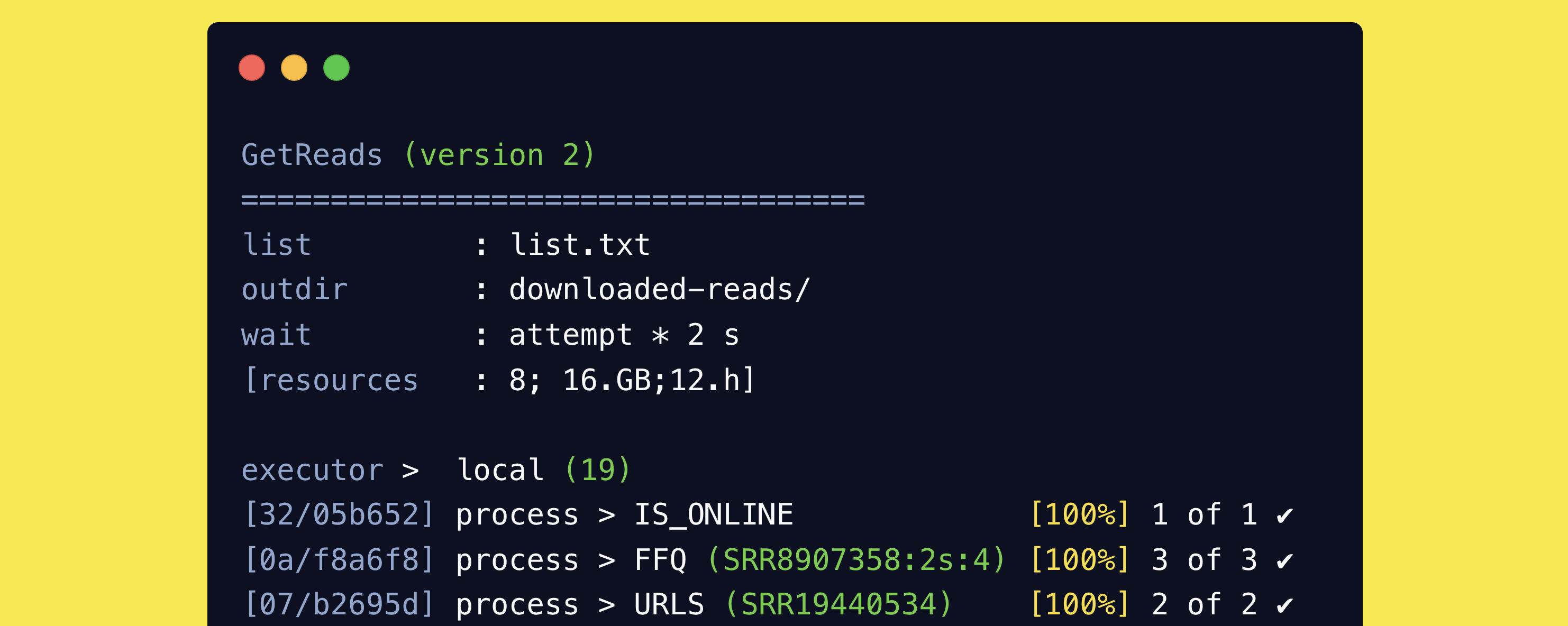
MetaPhage is a complete reads-to-report pipeline for viral metagenomics. It is a Nextflow pipeline that can be run on a local machine or on a cluster. It is designed to...
To identify putative viral sequences without using a classifier, we can use a de novo assembly approach. We first need to perform a standard metagenome assembly, then we need to...
This “virome primer” is designed to provide a short overview on viromics and the tools available to analyse viral metagenomes. The tutorial is agnostic to the environment, and can be...
“Virome refers to the assemblage of viruses that is often investigated and described by metagenomic sequencing of viral nucleic acids that are found associated with a particular ecosystem, organism or...
On our first day we well cover the concepts behind taxonomic classification using Kraken2 (and Bracken), and see how to remove host reads and perform the quality checks (and filtering)....
Kaiju can be installed, as usual, from Conda.
Like Kraken2, we have access to pre-built databases, and for this
tutorial we used the nr 2021-02-24 (52 GB).
When performing multiple operations on the same dataset, as some of you pointed out during the workshop, we often want to collate metadata. R is the ideal choice for doing...
To determine the microbial composition in your samples, one method to get this information is taxonomic read profiling. Here you compare your reads to a database of interest. Kraken2 is...
To determine the microbial composition in your samples, one method to get this information is taxonomic read profiling. Here you compare your reads to a database of interest. Kraken2 is...
In our workshop we provided a Kraken2 database for you to use. However, most of the time, you would need to create a database for your own host. For the...
Our goal here is to create a small and artificial set of sequences to be classified using Kraken2,
to practice with its parameters and its output formats.
First steps Log in into the server You should find a directory called sequences in your home. List the files in that directory (for example with ls -l ~/sequences) The...
If you are working with host-associate microbiome, we are usually only interested in the microbial fraction and not the host DNA. In fact, host DNA can intefere with the read...
Today we will use Braken to recalibrate our estimations, and a set of scripts to merge multiple samples in a single table, and see how to filter it. We will...
On our first day we well cover the concepts behind taxonomic classification using Kraken2 (and Bracken), and see how to remove host reads and perform the quality checks (and filtering)....
Deblur is an alternative method to produce
a set of denoised sequences. While DADA2 can
natively support paired-end reads, Deblur can
only manage single-end reads.
The idea to check if some condition is true before executing some commands is very simple and useful. The Bash syntax, for historical reasons, is probably the ugliest you’ll ever...
After a small introduction to Bash scripting, we finally create a first bioinformatics script… introducing one of the loops we can use with the shell. A loop is a structure...
After using the Linux terminal for a while, everyone wants to be able to write simple “scripts” to perform repetitive tasks. They looks like recipes, with the main difference of...
Deblur is an alternative method to produce
a set of denoised sequences. While DADA2 can
natively support paired-end reads, Deblur can
only manage single-end reads.
Deblur is an alternative method to produce
a set of denoised sequences. While DADA2 can
natively support paired-end reads, Deblur can
only manage single-end reads.
On our first day we well cover the concepts behind taxonomic classification using Kraken2 (and Bracken), and see how to remove host reads and perform the quality checks (and filtering)....
Kaiju can be installed, as usual, from Conda.
Like Kraken2, we have access to pre-built databases, and for this
tutorial we used the nr 2021-02-24 (52 GB).
When performing multiple operations on the same dataset, as some of you pointed out during the workshop, we often want to collate metadata. R is the ideal choice for doing...
To determine the microbial composition in your samples, one method to get this information is taxonomic read profiling. Here you compare your reads to a database of interest. Kraken2 is...
To determine the microbial composition in your samples, one method to get this information is taxonomic read profiling. Here you compare your reads to a database of interest. Kraken2 is...
In our workshop we provided a Kraken2 database for you to use. However, most of the time, you would need to create a database for your own host. For the...
Our goal here is to create a small and artificial set of sequences to be classified using Kraken2,
to practice with its parameters and its output formats.
First steps Log in into the server You should find a directory called sequences in your home. List the files in that directory (for example with ls -l ~/sequences) The...
If you are working with host-associate microbiome, we are usually only interested in the microbial fraction and not the host DNA. In fact, host DNA can intefere with the read...
Today we will use Braken to recalibrate our estimations, and a set of scripts to merge multiple samples in a single table, and see how to filter it. We will...
On our first day we well cover the concepts behind taxonomic classification using Kraken2 (and Bracken), and see how to remove host reads and perform the quality checks (and filtering)....
MetaPhage is a complete reads-to-report pipeline for viral metagenomics. It is a Nextflow pipeline that can be run on a local machine or on a cluster. It is designed to...
To identify putative viral sequences without using a classifier, we can use a de novo assembly approach. We first need to perform a standard metagenome assembly, then we need to...
This “virome primer” is designed to provide a short overview on viromics and the tools available to analyse viral metagenomes. The tutorial is agnostic to the environment, and can be...
“Virome refers to the assemblage of viruses that is often investigated and described by metagenomic sequencing of viral nucleic acids that are found associated with a particular ecosystem, organism or...
This “virome primer” is designed to provide a short overview on viromics and the tools available to analyse viral metagenomes. The tutorial is agnostic to the environment, and can be...
To identify putative viral sequences without using a classifier, we can use a de novo assembly approach. We first need to perform a standard metagenome assembly, then we need to...
MetaPhage is a complete reads-to-report pipeline for viral metagenomics. It is a Nextflow pipeline that can be run on a local machine or on a cluster. It is designed to...
Counting the number of occurrences of each word in a text is a common task in computational linguistics. It is also a good example of how to use Python to...
Counting the number of occurrences of each word in a text is a common task in computational linguistics. It is also a good example of how to use Python to...
Counting the number of occurrences of each word in a text is a common task in computational linguistics. It is also a good example of how to use Python to...
This walkthrough can be completely performed on any Linux terminal where Miniconda/Mamba have been installed, including a web based terminal offered by CLIMB BIG DATA new notebooks.