
SAM format

Here we introduce the SAM format, to store the result of a mapping of one or more DNA sequences against a reference genome
Mapping our first (small) dataset
Gathering the tools
We can start creating a dedicated environment for this tutorial:
1
2
3
mamba create -n learn_sam --yes -c bioconda -c conda-forge \
"samtools>=1.12" "bwa>=0.7.17" "seqfu>=1.11"
conda activate learn_sam
Creating a working directory
We will store our files in a dedicated directory:
1
2
mkdir -p ~/learn_sam
cd ~/learn_sam
We will start indexing the reference genome. This is a one-time operation, which will allow us to map reads against the reference genome as many times as we want.
1
bwa index ~/learn_bash/phage/vir_genomic.fna
Create a single sequence file
It’s tempting to try to aling a lot of short reads, after all this is the real-world scenario we are preparing for. But it’s even more important to remember that we need to be able to test if a tools does what we think it does.
We can create a single synthetic read from the reference genome.
Using only UNIX tools, we create a header with echo and then append the first line of the reference sequence (80 bases):
1
2
3
# Create a header and append a sequence
echo ">my-read" > my-read.fa
head -n 2 ~/learn_bash/phage/vir_genomic.fna | tail -n 1 >> my-read.fa
Alternaatively, we can use SeqFu cat to take a fraction of the sequence of known starting point and length.
We will use SeqFu head first to select the first sequence only (in our case we only have one, but for other exercises this will be useful).
Then with SeqFu cat we can:
-
--trim-front INTremove the first INT bases, thus selecting the sequence starting from the INT-th base -
--truncate INTprint a sequence of INT bases total (this will be the read length) -
-z(or--strip-name) will remove gthe original name -
-p(or--prefix STRING) will add a prefix to the name of the sequence (in our case, simply read)
1
2
seqfu head -n 1 ~/learn_bash/phage/vir_genomic.fna | \
seqfu cat --trim-front 1200 --truncate 100 -z -p read > read-fwd.fa
Now we can reverse complement it with SeqFu rc
(and then use SeqFu cat again to append, with -a STRING,a suffix to the read name),
and create a minimal reads file with a sequence and its reverse complement:
1
2
seqfu rc read-fwd.fa | seqfu cat -a "rc" > read-rc.fa
cat read-fwd.fa read-rc.fa > reads.fa
Mapping the reads
The general syntax when mapping reads with bwa is:
1
2
# We need to change the command accordingly
bwa mem $indexed_reference $short_read > file.sam
Where:
-
$indexed_referenceis the reference genome, indexed withbwa index -
$short_readis a FASTA file with the reads to map. If you have paired end reads, supply the R1 file first and then the R2 file. -
file.samis where the output will be redirected to
We can map our synthetic read against the reference genome:
1
bwa mem ~/learn_bash/phage/vir_genomic.fna reads.fa > my-alignment.sam
![]() Inspect the output file with
Inspect the output file with less -S.
A simplified view of the output:
1
2
3
@SQ SN:NC_001416.1 LN:48502
read1 0 NC_001416.1 1201 60 100M
read1rc 16 NC_001416.1 1201 60 100M
confirming that:
- the header will list all the reference sequences (SN), and their length (LN)
- our read was selected to start at 1200
- it’s 100 bases long: the CIGAR is 100M (100 matches) see later
- the first read has flag 0, the second 16 (match in the reverse strand) see later
The SAM format
![]() For a full description see the SAM format specification.
For a full description see the SAM format specification.
The SAM format is a tabular format, with one line per read. At the beginning there is a header, a set of
lines starting with @. The header contains information about the reference genome (one line per chromosome),
plus other informations about the mapping.
Each following line is a tab separated list of values, with the following fields:
| Field position | Field name | Description |
|---|---|---|
| 1 | QNAME |
Query template name (read name) |
| 2 | FLAG |
bitwise FLAG |
| 3 | RNAME |
Reference sequence name (chromosome name in the reference) |
| 4 | POS |
1-based leftmost mapping position |
| 5 | MAPQ |
Mapping Quality |
| 6 | CIGAR |
CIGAR string |
| 7 | RNEXT |
Ref. name of the mate/next read |
| 8 | PNEXT |
Position of the mate/next read |
| 9 | TLEN |
observed Template length (insert size) |
| 10 | SEQ |
segment SEQuence (the read, excluding the clipped bases) |
| 11 | QUAL |
ASCII of Phred-scaled base QUALity+33 (the read’s quality string, excluding the clipped bases) |
There are two complicated fields we will just briefly mention:
-
FLAGis a bitfield, with each bit representing a different property of the read. It’s a set of boolean properties, each being true or false, and the sum of them is the value of the field. For example, if the read is mapped in the reverse strand, the 5th bit is set to 1, and the total value of the field is 16. See this page for a full description of the flags. -
CIGARis a string describing the alignment of the read against the reference. It’s a list of operations, each operation is a number followed by a letter. The number is the length of the operation, and the letter is the type of operation. See this page for more details.
Converting to BAM
The SAM format is a text format, and it’s not very efficient.
We can convert it to a binary format, BAM, with
the samtools view command. Samtools view is used both to convert a text SAM file
to a BAM file (when specifying -b), and to convert a BAM file to a text SAM file.
There are several options which allows to filter the alignments by location and other attributes. We will not cover them here, but you can find more information in the Samtools manual.
1
samtools view -b my-alignment.sam > my-alignment.bam
The output is no longer a text file!
Sorting the BAM file
Several programs require the BAM file to be sorted and indexed. The index is a tiny file that
works exactly as the index of a book: it allows you to jump to a specific position in the file.
We can sort it with samtools sort and create the index with samtools index:
![]() We will add
We will add \ to split commands over multiple lines
1
2
3
4
5
6
7
8
9
10
# Sort by reference name and then position
samtools sort \
-o my-alignment.sorted.bam \
my-alignment.bam
# Create the index
samtools index my-alignment.sorted.bam
# What did the indexing step create?
ls -ltr my-*
We can create the BAM file, sort and index it in one step, using pipes.
It’s a common convention for programs to read the standard input (i. e. from a pipe)
when the input file is specified as -.
1
2
3
samtools view -b my-alignment.sam | \
samtools sort --write-index \
-o pipe-sorted.bam -
![]() Try to alter the input file with insertions, deletions or mismatches.
Try to alter the input file with insertions, deletions or mismatches.
Aligning a full dataset
Using our “learn_bash” repository we can create a BAM file with a full dataset.
In the phage subdirectory we have the reference genome we just used, and a subdirectory called reads, which contains 4 samples of reads, each with 2 files, one for the forward reads and one for the reverse reads (“R1” and “R2”, respectively).
We already indexed the genome so we can directly align the reads.
First, let’s move to the phage directory and create a dedicated directory for the output files
1
2
cd ~/learn_bash/phage/
mkdir mapped
Let’s count the number of reads in the datasets (seqfu count will identify the paired datasets and report the number of fragments):
1
seqfu count reads/*.fastq.gz
Then we can map one sample (we will use sample2), this time directly with a pipe:
1
2
bwa mem vir_genomic.fna reads/sample2_R1.fastq.gz reads/sample2_R2.fastq.gz | \
samtools sort --write-index -o mapped/sample2.bam -
![]() We skipped the
We skipped the view step, because samtools sort can also convert from SAM to BAM.
A first overview
We can use samtools stats to get a first overview of the alignment.
It’s output is very long, so we will pipe it to less -S to make it more readable.
1
samtools stats mapped/sample2.bam | less -S
![]() How many reads are mapped according to this report? Is this consistent with
the output of
How many reads are mapped according to this report? Is this consistent with
the output of seqfu count?
![]() How many reads are “reads properly paired”? What does this mean?
How many reads are “reads properly paired”? What does this mean?
We can load the reference genome and the indexed BAM file in IGV to have a look at the alignments:
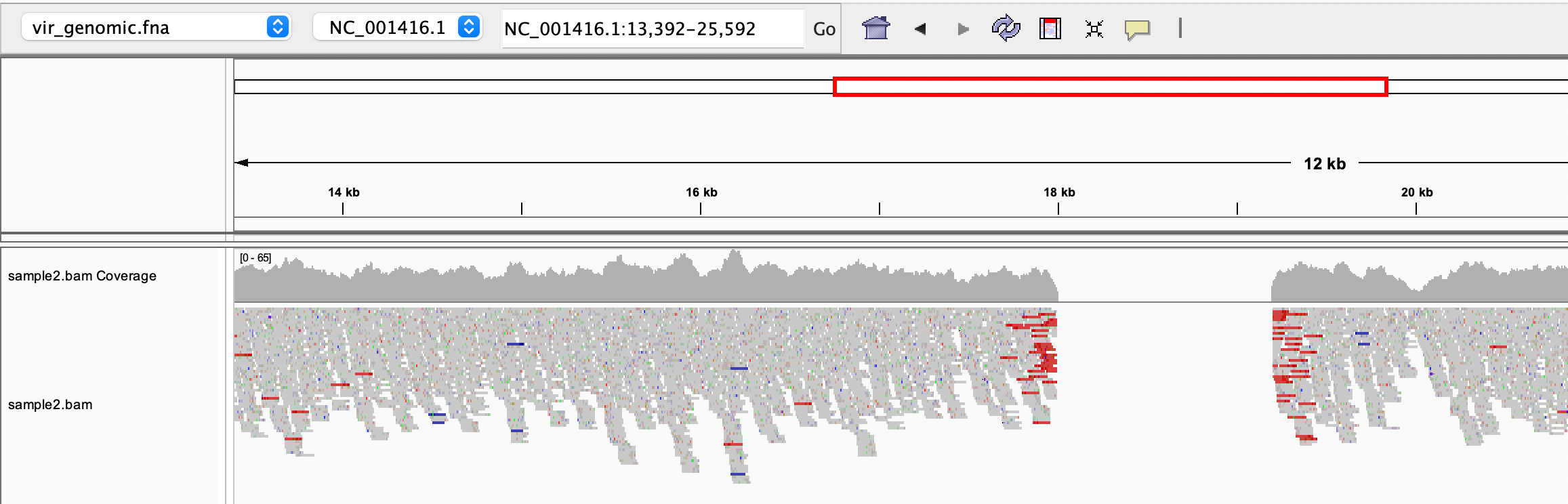
Can you spot the deletion? Note that the paired reads are coloured in red because they map on the reference with a larger than expected insert size (i. e. in the genome we sequenced, a portion of the reference is missing)