Overview
In bioinformatics, a common task is to align several (usually short) DNA sequences against a reference sequence (e. g. a complete genome of the organism).
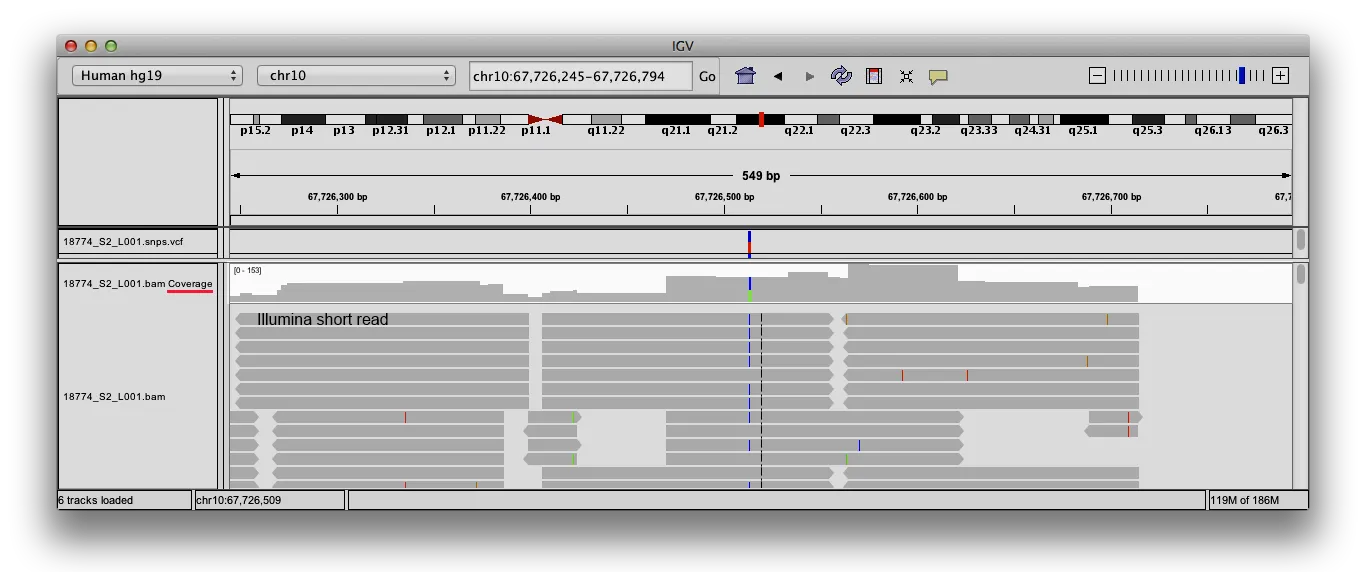
The screenshot of shows the alignment of several short DNA sequences (reads) against a reference genome. The program graphically displays the coverage track
IGV itself displays a coverage track, but for computational reasons only if the zoom is below 50 kbp. On the other hand, it is possible to visualize a BED track spanning the whole chromosome.
bamtocov reads an alignment file in BAM format and prints a BED track with the nucleotide coverage and can be used to select the regions with a coverage of interest (e. g. uncovered regions with 0X coverage, low coverage regions <5X, etc.).
Features
✔️ can read sorted BAM files even without the index
✔️ can read sorted BAM files from streams (from a pipe)
✔️ can calculate the physical coverage to check the assembly integrity
✔️ can calculate the per-strand coverage to check for strand bias
✔️ lowest memory usage for a coverage tool
File formats
BED files
A BED file (.bed) is a tab-delimited text file that defines a feature track. In this context the magnitude refers to the nucleotide coverage of the interval.
The columns are chromosome name, start position (inclusive, zero-based), end position (non-inclusive, zero-based) and coverage. An example is:
seq1 0 9 0
seq1 9 109 5
seq1 109 189 0
seq1 189 200 2
Target statistics
⚠️ this format is not final.
For each sample, 5 columns are printed:
bam_basesbam_meanbam_minbam_maxbam_length
| interval | bam_bases | bam_mean | bam_min | bam_max | bam_length |
|---|---|---|---|---|---|
| target1_8X | 699 | 3.495 | 1 | 6 | 200 |
| target2_0X | 0 | 0.0 | 0 | 0 | 50 |
| target3_1X | . | . | . | . | . |
| for_rev_10Xa | 100 | 10.0 | 10 | 10 | 10 |
| for_rev_10Xb | 100 | 10.0 | 10 | 10 | 10 |
| for_rev_10Xc | . | . | . | . | . |
Further reading
For a concise introduction to sequence and physical coverage see: What is sequence coverage?.