
Get FASTQ reads from NCBI: an automated workflow
The problem
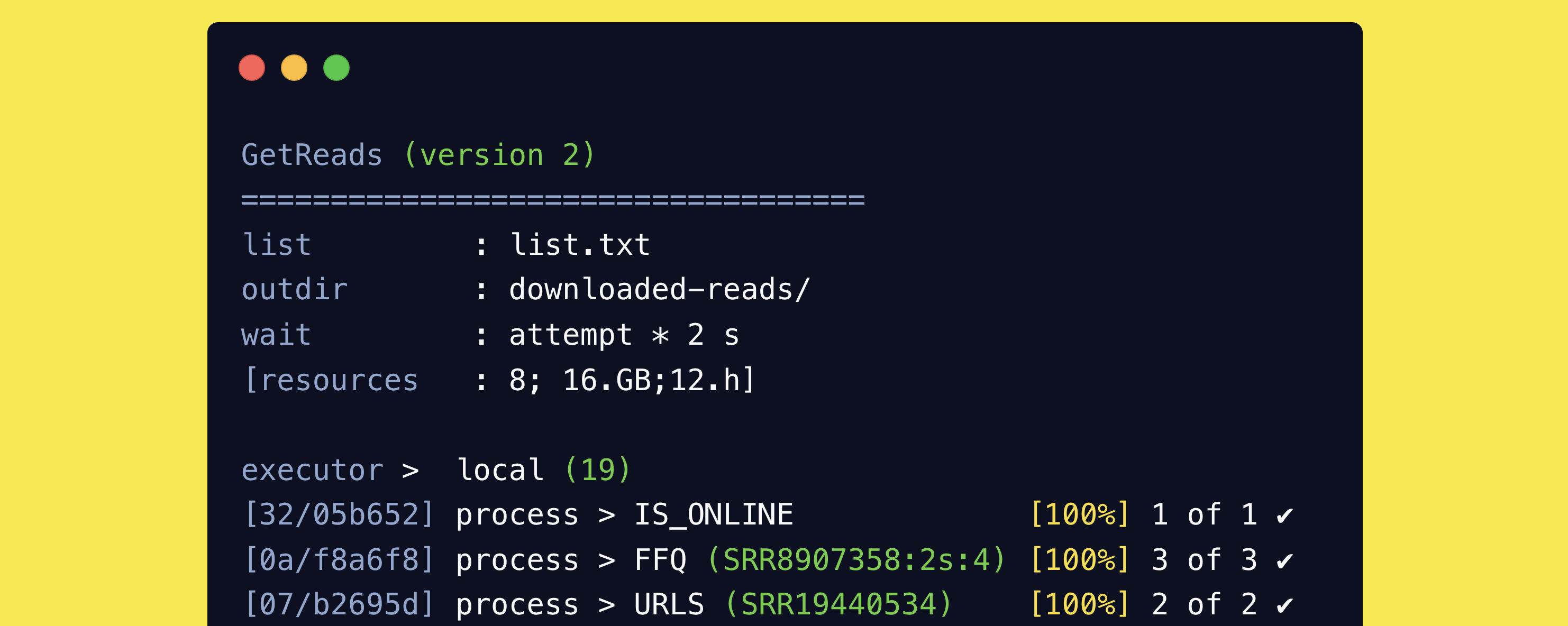
The NCBI SRA database is a treasure trove of sequencing data, but it is not always easy to download the data you need. I n this post we will see how to automate the download of FASTQ files from the SRA database, using a Nextflow pipeline called getreads.
Being written in Nextflow it can handle for us:
- the dependencies (either via conda or docker)
- the processes (tasks can run in parallel and even on a distributed cluster if available)
The solution
The getreads pipeline is available on GitHub but can be installed automatically via Nextflow.
What you need is a machine with:
- Nextflow installed (can be installed via conda)
1
conda install --yes -c bioconda -c conda-forge nextflow
- Either conda or docker installed, to handle the dependencies
- Try typing
docker --versionto check if you have docker installed: it’s the preferred way to run the pipeline - Try typing
conda --versionto check if you have conda installed: it’s the fallback option if you don’t have docker
- Try typing
How to use the pipeline
- Create a text file with a list of SRA accessions (one per line), for example:
1
2
SRR19440534
SRR19440543
- Run the pipeline like:
1
nextflow run telatin/getreads -r main --list list.txt -profile docker
This step - when run for the first time - will download the pipeline and all the dependencies,
and then will execute the pipeline on the data in list.txt.
The pipeline will create a folder called getreads with the pipeline outpout files (can be changed
passing the --outdir DirectoryName option). A subfolder called reads will contain the FASTQ files.
![]() If you don’t have Docker installed, type
If you don’t have Docker installed, type -profile conda instead
Other parameters
The pipeline can be customized with the following parameters:
-
--max_cpus: maximum number of CPUs to use (default: 8) -
--max_memory: maximum amount of memory to use (default: 16 GB)
See the GitHub page for more information