What is sequence coverage?
Sequence coverage, or sequencing depth, is the number of aligned reads that include a given nucleotide of a reference genome. It is usually displayed as a coverage track above the read alignments: peaks indicate heavily sequenced positions, while drops highlight poorly supported regions.
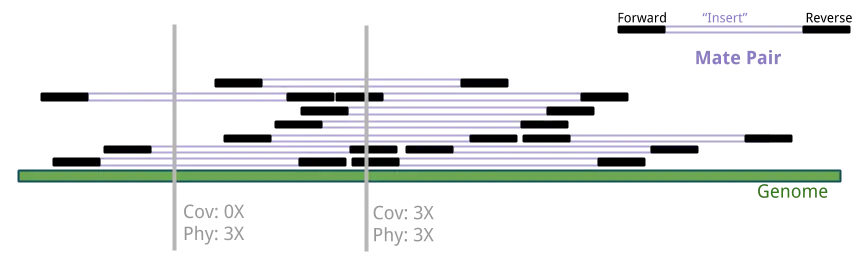
Sequence coverage counts bases directly covered by reads. Physical coverage counts bases covered or spanned by paired-end fragments.
Sequence coverage and physical coverage
With paired-end or mate-pair libraries, sequence coverage and physical coverage answer different questions:
- Sequence coverage asks how many reads include a base.
- Physical coverage asks how many fragments span a base.
This distinction matters when the bases in a region are not sequenced uniformly, but paired reads still connect the surrounding sequence. BamToCov can report both measures.
From coverage to regions
Coverage tracks become more useful when they are converted into intervals. Instead of inspecting one nucleotide at a time, you can collapse the profile into BED features and mark regions above or below a threshold.
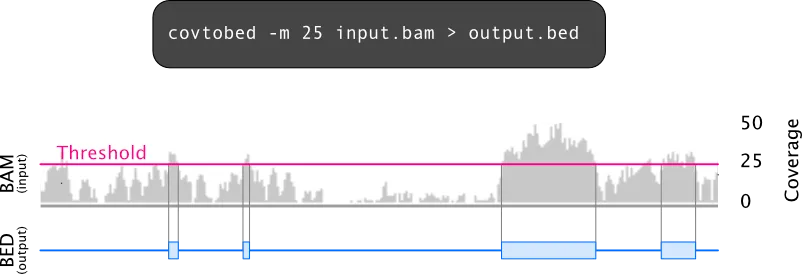
A coverage profile can be summarized as BED intervals, for example to report regions above or below a chosen threshold.
This is useful to identify:
- uncovered regions
- low-depth intervals
- unexpectedly high coverage
- targets meeting a minimum depth requirement
Coverage and misassemblies
Coverage is also a diagnostic signal. A contig may show continuous sequence coverage while the physical coverage drops sharply, suggesting that paired reads no longer bridge the region consistently.
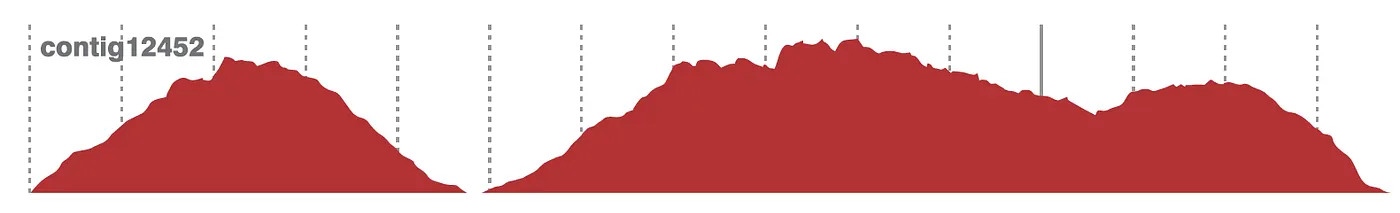
A sharp drop in physical coverage can reveal a chimeric or misassembled contig even when sequence coverage looks continuous.
Why it matters
Genome browsers such as IGV are excellent for inspecting small loci, but coverage tracks are easier to review genome-wide once exported as BED. This is the main workflow supported by BamToCov: read alignments from BAM, compute sequence or physical coverage, and summarize the regions that matter.